Purpose
Here, we describe the design of a modular CRISPR landing pad installed at a Chlamydomonas reinhardtii safe-harbor locus. The landing pad will be engineered as a reusable genomic “port” that provides a fixed chromosomal environment for high-level, consistent nuclear expression of hundreds to thousands of genetic variants. Our long-term goal is to build a C. reinhardtii chassis to serve as a scalable testbed for deep mutational scanning, engineered protein variant screening, optogenetic discovery, and multi-gene pathway refactoring.
The landing pad will contain a removable stuffer cassette encoding a fluorescent protein and selectable marker, flanked by short synthetic homology arms and embedded within a stable promoter-intron-terminator expression framework. The landing pad will be installed at a safe-harbor locus in a host strain with srta-attenuated transgene silencing. New payloads will be introduced through a simple, repeatable HDR-mediated cassette swap driven by Cas9 RNP excision of the stuffer. In this workflow, landing pad installation will be a one-time, QC-intensive genome-engineering step, while subsequent payload insertion will be a high-throughput, QC-light operation. This separation will decouple strain construction from variant deployment, enabling consistent nuclear expression and scalable installation of hundreds to thousands of pooled genetic payloads in a fixed chromosomal context.
We outline a conceptual and technical blueprint for this system: why C. reinhardtii needs a universal expression port, how we'll build it, how it'll work mechanistically, and how the two-stage workflow enables variant libraries at scales previously unrealistic in this organism. This work presents a design and implementation blueprint rather than a completed system, integrating recent advances into a unified framework that we aim to build and validate iteratively with community input. We’re sharing this design now to gather early community input. We welcome feedback on both the overall approach and specific design choices.
Motivation
For decades, C. reinhardtii has been a workhorse of plant and ciliary biology. In many ways, it has been recognized as the plantae counterpart to Saccharomyces cerevisiae 1–2. It packages fundamental plant cellular biology into a unicellular, easy-to-culture microbe with well-defined, quantifiable phenotypes in vegetative haploid and diploid life cycles. It swims, senses light, builds and resorbs cilia, and operates a chloroplast — all within a single, tractable nuclear genome context. This makes C. reinhardtii invaluable for studying processes such as photosynthesis, ciliary biology, chloroplast-nuclear communication, opsin structure-function, and a wide range of evolutionary questions accessible through Zoogle (Arcadia’s comparative genomics database 3).
However, unlike S. cerevisiae, synthetic biology toolkits for C. reinhardtii remain fragmented, largely due to clade and organism-specific obstacles. Transformation efficiency is highly variable and homology-directed repair (HDR) is rare. Non-homologous end joining (NHEJ) is the favored mode of repair in most nuclear editing attempts 4–6. Silencing pathways selectively shut down foreign promoters and exogenous coding sequences, making robust nuclear expression of transgenes unusually fragile 7. Although NHEJ does facilitate nuclear integration, random insertion places transgenes into highly variable chromatin environments. Identical constructs integrated at different loci routinely show order-of-magnitude differences in expression, driven by local transcriptional activity, epigenetic state, and silencing susceptibility 4–6, 8. This locus-dependent variability makes direct comparison between integrants unreliable and forces extensive clone screening, normalization, or ad hoc selection of representative lines. Moreover, random integration requires whole-genome sequencing or locus-mapping PCR to identify insertion sites and copy number variation. Together, these constraints severely limit scalability and interpretability, especially for variant libraries and quantitative structure-function analyses. This has limited the overall ease of effective transgene knock-ins in C. reinhardtii and the degree to which the field can scalably express large variant libraries, the backbone of structure-function analyses and protein engineering.
In contrast, the concept of a reusable genomic port or landing pad is now common in yeast and mammalian cells: choose a safe-harbor locus, integrate a standardized expression site, and swap with new cassettes iteratively and indefinitely 9–12. In C. reinhardtii, we have simply lacked a combination of (i) a validated safe harbor, (ii) reliably high HDR, and (iii) a unified cloning and donor format, until relatively recently.
The landscape for genome engineering in C. reinhardtii has changed dramatically in the last five years. Cell synchronization through light-dark cycling has been shown to increase nuclear transformation and HDR by enriching the population in HDR-competent phases of the cell cycle 4. Transformation with purified Cas9 ribonucleoproteins (RNPs), rather than plasmid-expressed nucleases, has been shown to reliably enhance nuclear gene disruption and targeted insertional mutagenesis, especially when coupled with robust, enzymatic cell wall removal 5. The use of short donor homology arms (~50 bp) with clean linear templates have been shown to support efficient, precise HDR at endogenous genes 5–6, 13. Labs have been leveraging this to develop efficient, standardized workflows and toolkits for CRISPR design, execution, and validation 5–6, 13. Further, Sir2-type histone deacetylase (SRTA), the main C. reinhardtii sirtuin-like histone deacetylase (HDAC), has been identified as largely responsible for transgene silencing 7. Lastly, several loci have been investigated as potential safe harbor loci; notably, the LHCBM1 locus (Cre01.g066917) tolerates targeted integration, exhibits consistently high transcription, and supports strong, stable expression when used as a fusion target 8. Taken together, this presents an opportunity to build a single, well-characterized genomic gateway to minimize the effort and variability required to introduce transgenes into C. reinhardtii. Here, we're not introducing new editing chemistry. Instead, we're consolidating existing advances into a reusable, strain-level infrastructure designed to support library-scale experiments.
The idea
We aim to install a landing pad that provides a stable, predictable, high-expression environment for a wide variety of future payloads. Our design encompasses the landing pad strain and examples of payload cassettes — and addresses three bottlenecks at once: positional variability, unpredictable silencing, and the inefficiency of designing new donors and QC assays for each experiment. This design features a defined chromosomal site within a specific genetic background, a standardized expression cassette, a removable stuffer module, and a set of engineered homology boundaries that allow us to slot in new inserts, akin to a plasmid multiple-cloning site, enabling easy CRISPR-mediated HDR insertion of future genes of interest (GOI) or gene variants.
The design philosophy behind this system reflects the practical constraints of C. reinhardtii genome editing and the demands of scalable protein engineering. We prioritize infrastructure over one-off edits: consistency over scarlessness, engineered interfaces over ad hoc cloning, and long-term strain reusability over bespoke optimization.
Landing pad insertion — Genetic background and safe harbor locus
A major obstacle in C. reinhardtii engineering has been extreme transcriptional silencing of exogenous material. More recently, SRTA-mediated H3K9 methylation has been identified as the major causal driver, with subsequent chromatin compaction occurring at exogenous promoter architectures and coding sequences that diverge from native nuclear genes, including viral promoters, bacterial ORFs, synthetic constructs, and heterologous, codon-biased ORFs lacking C. reinhardtii introns 7. Loss-of-function srta mutants dramatically reduce this repression: inserted, intronless ORFs with exogenous promoters remain transcriptionally active, with stable expression maintained across many generations 7. Native loci and endogenous gene expression remain unaffected, and srta mutants show no measurable defects in growth, photosynthetic performance, or stress responses. Other factors (e.g., DMC5, VIG1) can further enhance transgene expression, but act through broader regulatory pathways and have more significant global effects on fitness and the rest of the genome 14. SRTA disruption provides the cleanest, most transgene-specific relief from epigenetic silencing. For this work, we’ll leverage a srta-attenuated background as a stabilizing foundation rather than as a strict requirement, layering it with additional architectural safeguards that promote robust expression even in partially silencing-competent strains. With silencing risk reduced, the next major constraint is positional variability.
To ensure stable nuclear expression in this silencing-attenuated background, the landing pad will be installed in a permissive genomic locus, a safe harbor that can support predictable, long-term transgene activity. Multiple lines of evidence establish LHCBM1 as such a locus 8. In a systematic comparison of transcriptionally active, non-essential candidate sites, including PPX1, ARS2, FAP138, FAP206, Cre03.g196600, and Cre06.g278750, LHCBM1 consistently produced the strongest and most uniform reporter expression, the lowest clone-to-clone variability, and the highest frequency of correct CRISPR-mediated HDR integration 8. These loci were selected based on standard safe-harbor criteria: high endogenous transcription, sufficient intergenic space, and lack of locus-linked silencing. Expression from LHCBM1-targeted knock-ins remained stable for at least 18 months of continuous culture without detectable silencing, and the locus supported robust functional bioproduction (terpene synthase activity), yielding the highest metabolite titers of all sites tested 8. LHCBM1 encodes a subunit of the major light-harvesting complex II (LHCII), a photosynthetic antenna protein that exhibits strong, consistent expression under standard vegetative conditions. This sustained transcription is consistent with an open chromatin environment that's permissive for Cas9 access and homology-directed repair 8. Consistent with principles of transcription-coupled DNA repair, double-strand breaks within actively transcribed genes rapidly accumulate γH2AX, recruit homologous recombination machinery, and suppress NHEJ through 53BP1 antagonism 15. Previous strategies fused transgenes directly to the LHCBM1 coding sequence, either as C-terminal fusions or via viral 2A peptides, enabling bi-cistronic (cytosolic) expression of separate proteins 8. This yielded strong, light-responsive expression, but introduced modest fitness costs by perturbing LHCBM1’s native transcription and protein output. To avoid this, we’ll position the landing pad downstream of the LHCBM1 transcription unit, outside the annotated coding sequence and 3′ untranslated region, close enough to exploit the locus’s open chromatin environment while preserving native promoter activity, transcript processing, and gene function.
As a secondary control and backup integration site, PPX1 (Cre09.g396300), a phenotypically neutral nuclear gene encoding protoporphyrinogen IX oxidase, will be used as an alternative landing pad locus. PPX1 supports efficient CRISPR-mediated HDR and has been validated as a permissive insertion site, enabling direct, head-to-head comparison of expression, stability, and repair efficiency against LHCBM1 in a distinct chromatin context 8.
After installing the landing pad and subsequent payloads, we will validate growth, chlorophyll fluorescence (including non-photochemical quenching), and LHCBM1/payload transcript and protein levels (qPCR and Western blotting) across passages to ensure that neither integration perturbs cellular physiology. Minimizing fitness effects and maximizing expression consistency — robust and insensitive to environmental fluctuations — is key to comparing inserted GOIs and variants across different individuals, especially at scale. Inducibility can be tuned in the future by implementing modular inducible promoters once the landing pad is established at the site.
From a practical standpoint, the landing pad system is designed to be installed in a defined, swimming-competent wild-type background. We aim to start with CC-124, a cell-walled laboratory strain that retains robust flagellar motility and serves as a common reference for behavioral and phototaxis assays. CRISPR-mediated gene knockouts can be generated directly in this background. We will disrupt SRTA prior to landing pad installation to establish a silencing-attenuated chassis, while additional gene knockouts can be introduced either before or after landing pad insertion, depending on experimental needs. We have chosen selection markers that will remain orthogonal across editing stages: SRTA disruption uses nourseothricin resistance, the landing pad stuffer cassette confers blasticidin resistance, and payload insertion replaces the stuffer with zeocin resistance, enabling unambiguous selection at each step, and maintaining compatibility with common mutant collections (the hygromycin and paromomycin resistance markers used in the CLiP2 and CLiP1 mutant collections, respectively) 16–17. Although the landing pad architecture is compatible with genetic crosses or transfer into existing mutant libraries, such approaches necessarily introduce mixed genetic backgrounds and strain-specific constraints. For applications where sensitive phenotypes such as swimming, phototaxis, or fitness are primary readouts, direct CRISPR-mediated editing in a defined wild-type background should provide a cleaner, more scalable foundation for variant screening.
Together, these design choices should yield a landing pad chassis that's expression-robust and compatible with existing C. reinhardtii mutant resources. The system doesn't depend on any single component. It's intentionally modular, so it can operate across genetic backgrounds while layering in additional safeguards when expression stability matters most.
Conceptual architecture of the landing pad
The landing pad itself is defined by four key features (Figure 1):
- A stable and silencing-resistant expression unit
- A removable double-reporter stuffer cassette
- Engineered synthetic homology arms
- Flanking CRISPR cut sites (protospacer–PAM modules)
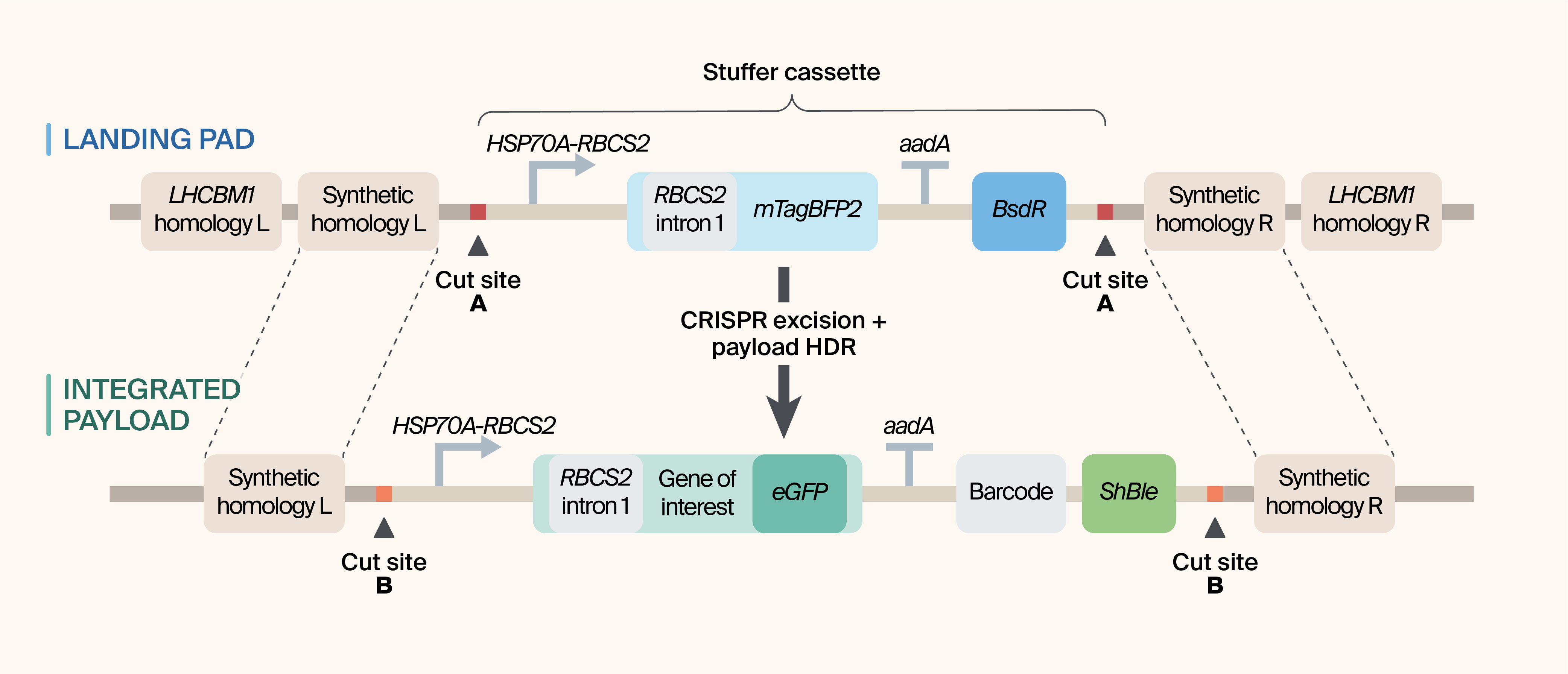
Figure 1. Landing pad and payload architecture for CRISPR-mediated cassette exchange at the LHCBM1 locus.
(Top) Genomically integrated landing pad showing LHCBM1-flanking homology arms, synthetic CRISPR cut sites (protospacer–PAM modules), and a removable stuffer cassette.
(Bottom) Payload HDR donor template used to replace the excised stuffer following Cas9 cleavage at the flanking cut sites. The shared synthetic homology arms and reusable CRISPR cut sites define a precise excision window, enabling iterative, locus-defined insertion of single or multi-cassette payloads with standardized junctions and streamlined quality control.
Promoter, intron, and terminator
Our proposed landing pad architecture minimizes silencing by using validated, endogenous-like regulatory elements (HSP70A-RBCS2 promoter, RBCS2 intron 1), a validated terminator (aadA), and compact, chromatin-neutral synthetic homology arms (Figure 1). The RBCS2 promoter alone is often weak and environmentally sensitive, but fusion to HSP70A yields a stronger, more stable promoter that maintains robust expression across fluctuating conditions 18. The RBCS2 intron 1 provides intron-mediated enhancement of expression (IME) and mimics native nuclear gene architecture 19. The aadA terminator has been demonstrated to function reliably at the LHCBM1 locus 8.
Synthetic homology arms
The design employs two sets of homology arms: an outer set homologous to a noncoding region downstream of the LHCBM1 locus for initial landing pad integration, and an inner set of fully synthetic homology arms used for all subsequent payload swaps (Figure 1). In both cases, we propose using ~50 bp homology arms, following short-arm HDR strategies shown to be highly effective in C. reinhardtii 5–6.
Because these synthetic homology arms define the universal docking interfaces for all future integrations, we’d engineer them with strict constraints:
- No internal Type IIS sites (BsaI, BsmBI, SapI), enabling seamless Golden Gate assembly
- Balanced GC content, improving both DNA synthesis and PCR performance
- Universal primer-binding regions positioned immediately outside each arm for standardized QC
- Minimal internal microhomology to suppress microhomology-mediated end joining in favor of precise HDR
By fixing these HDR interfaces across all payloads, integration QC is collapsed into a single reusable workflow that should scale cleanly from a small number of constructs to many thousands.
The stuffer cassette: FP and Bsd for double-negative QC
To enable rapid validation of correct HDR swapping at scale, the proposed landing pad contains a dual-marker stuffer cassette encoding a fluorescent protein (mTagBFP2) and a blasticidin-resistance gene (bsr; BsdR) (Figure 1). Each marker would be expressed from its own HSP70A-RBCS2 promoter with an RBCS2 intron 1 and aadA terminator, matching the regulatory architecture used for payloads. Stable expression enables straightforward detection of correct landing pad installation and subsequent stuffer removal. Partial HDR events that remove only one marker should generate distinctive FP+/Bsd− or FP−/Bsd+ phenotypes. Correct HDR swapping should yield a double-negative phenotype (FP−/Bsd−), providing a fast, high-confidence readout for proper cassette exchange.
Fluorescent marker selection (stuffer vs. payload)
To maximize robustness of swap calling on standard microscope hardware, the stuffer cassette uses a blue fluorescent protein (mTagBFP2), while payloads use green and/or red fluorophores (eGFP and mScarlet-I) (Figure 1). This choice reflects two constraints: (i) C. reinhardtii exhibits strong chlorophyll autofluorescence that can complicate red-channel interpretation, and (ii) the imaging workflow is limited to a small number of excitation lines and emission bandpasses. Reserving the blue channel for the disposable stuffer preserves flexibility for payload designs, including dual-fluorophore payloads, while maintaining unambiguous detection of stuffer loss during HDR-mediated cassette replacement 20–21.
Because the stuffer fluorophore is used exclusively for swap QC, higher-performance fluorophores are reserved for payloads. In practice, correct swaps can be identified as BFP-negative and blasticidin-sensitive colonies, with payload integration confirmed by zeocin resistance and appearance of the payload fluorophore(s). Stuffer loss can also be screened at the colony level using gel documentation or plate-imaging systems, but because blue fluorophores impose stricter excitation requirements, such assays are treated as coarse prescreens rather than definitive tests. Accordingly, antibiotic selection and microscopy remain the primary criteria for confirming stuffer excision, whereas green and red payload fluorophores are readily detectable on standard plate imagers and support robust post-swap screening 20–21.
Synthetic protospacers for precise excision
The stuffer cassette in our proposed design is flanked by two synthetic CRISPR cut sites (protospacer–PAM modules) positioned exactly at the boundaries of the internal synthetic homology arms (Figure 1). During a payload swap, Cas9 RNPs would cleave both landing pad cut sites, excising the stuffer as a single contiguous fragment and creating a well-defined gap that will be repaired in one HDR event using a linear donor. This two-cut strategy enforces complete replacement of the stuffer rather than partial insertion or one-sided extension, reducing stuffer carryover and hybrid architectures that can arise in single-cut designs.
Each cut site consists of a 20 bp protospacer followed by an NGG PAM embedded in engineered sequence absent from the C. reinhardtii genome and from payload ORFs, enabling consistent on-target cutting across swaps (Figure 1). Within any given landing pad installation, the same protospacer sequence will be included on both sides of the stuffer cassette, allowing excision with a single sgRNA and supporting symmetric cleavage and efficient HDR. Because this protospacer pair is fixed at the landing pad level, the same guide RNA can be reused for all future payload replacements at this locus, independent of payload identity. Within a given landing pad configuration, a single sgRNA is reused for all swaps; alternative protospacer pairs are only introduced when designing re-entrant or multi-cycle architectures. Across payload designs, alternative protospacer–PAM pairs can be intentionally used (e.g., cut site A vs. cut site B; Figure 1) to support iterative replacement, while each individual design remains flanked by a matched pair of identical sites.
Following excision and HDR, the locus is effectively sealed against re-cleavage unless new cut-site modules are intentionally introduced. Iterative (“re-entrant”) swapping is therefore controlled at the payload level: payload backbones can reintroduce synthetic cut sites adjacent to the homology arms to enable further targeted replacement or refinement in derived strains without disturbing the original landing pad boundaries. Conversely, payloads that omit such cut sites should permanently seal the locus, which may be preferred when a final genetic configuration is reached or when additional CRISPR edits are planned elsewhere in the genome.
Conceptual architecture of the payload inserts
The proposed CRISPR landing pad system supports a variety of payload structures. Donor templates may encode a single expression unit or multiple linked expression cassettes, provided they are contained within the homology boundaries and delivered as a single linear repair template. Payloads can therefore be configured in multiple ways, including direct GOI-fluorescent protein fusions, bicistronic GOI-2A-fluorescent protein designs, or co-expression of a GOI and fluorescent protein with distinct regulatory elements. This enables co-insertion, for example, of a fluorescent organelle marker together with a wild-type rescue allele or reporter, supporting combinatorial or internally controlled experiments from a single, locus-defined integration. Further, payload ORFs can optionally include subcellular targeting signals or localization motifs, enabling standardized localization assays within a fixed genomic context. Although increasing donor size is expected to reduce absolute HDR frequency, antibiotic selection following stuffer excision provides strong biological enrichment for correct integrants, making multi-cassette payloads practical for targeted and moderate-scale applications. Payload size is therefore flexible by design, constrained primarily by HDR efficiency rather than by landing pad architecture.
By default, payloads use the same HSP70A-RBCS2 promoter and RBCS2-derived 5′UTR/intron architecture as the landing pad to maximize generality and comparability across variants. However, the modular design permits substitution of alternative 5′UTRs when translational control is limiting. In particular, βTUB2-derived 5′UTRs can be employed for payloads prone to leaky scanning, aberrant start-site selection, or N-terminal truncation, without altering chromatin context, genomic position, or HDR-based integration logic.
Payloads include a flexible linker between the ORF and tag. Fluorescent tags (mScarlet-I and eGFP) support localization assays and rapid FACS-based enrichment, while small epitope tags (HA, FLAG, Myc, His) minimize steric disruption during functional screens. Each payload also carries shBle, driven by the PSAD promoter, intron, and terminator, enabling zeocin selection of correct HDR integrants after stuffer excision — a critical biological amplifier of rare HDR events, particularly in pooled screens 5.
For pooled experiments, payloads incorporate a 12–20 bp non-transcribed barcode positioned downstream of the terminator and upstream of the ShBle antibiotic resistance cassette. This placement should ensure the barcode is genetically stable, non-expressed, and uniform across constructs, while remaining easy to PCR from crude genomic DNA for sequencing-based quantification and isolated from expression effects on the protein of interest, consistent with established C. reinhardtii toolkits 5–6. In pooled experiments, barcode abundance can serve as a proxy for payload representation, enabling sequencing-based tracking of variant fitness or enrichment without direct measurement of the encoded protein.
Cloning and donor assembly
Landing pad donors and the universal donor backbone can be prepared using traditional cloning and DNA synthesis, whereas payload insert donors can be generated using a defined Golden Gate assembly strategy. Previous modular cloning toolkits for Chlamydomonas enabled rapid construct assembly but relied on random nuclear integration, necessitating population-averaged expression measurements to mitigate strong locus effects 22. By contrast, the landing pad strategy described here fixes genomic context, enabling direct, quantitative comparison of variants.
Golden Gate cloning uses Type IIS restriction enzymes, which cut outside their recognition sites and enable scarless assembly with intrinsic error correction. Correctly assembled products eliminate internal Type IIS sites and are no longer cut, whereas misassembled intermediates retain these sites and are repeatedly digested and re-ligated. This self-correcting cycle yields a more uniform, high-fidelity donor pool, reducing the prevalence of aberrant constructs that could compromise downstream HDR precision 4–6.
The assembly workflow we propose begins with a standardized donor backbone containing the promoter, intron, terminator, synthetic homology arms, CRISPR cut sites, barcode, and shBle selection cassette. This modular architecture should allow thousands of ORFs to be pooled or assembled in 96- or 384-well arrayed formats using automated liquid handling. A single backbone can be assembled with diverse ORF libraries in one Golden Gate reaction. ORFs can be rapidly generated at scale with gene synthesis such that each is codon-optimized, contains an RBCS2 intron, and is flanked by Golden Gate BsaI sites. BspQI linearization followed by SPRI bead purification yields arrayed or pooled linear donor DNA suitable for effective HDR integration in C. reinhardtii 5–6. Donor DNA must be free of residual backbone, endotoxin, contaminants, and nicked species, as these have been shown to be critical determinants of HDR efficiency in C. reinhardtii 4–6.
RNP design and assembly
In C. reinhardtii, where nuclear editing windows are short and stress sensitivity is high, pre-assembled Cas9 RNP complexes act immediately upon delivery and clear rapidly, creating a narrow but consistent window during which HDR is favored 5–6. The purity, stoichiometry, and handling of Cas9 RNPs, and guide RNAs have been repeatedly identified as dominant variables of editing success in this organism 5–6. Accordingly, we propose using high-purity commercial Cas9 nuclease, stored in single-use aliquots at −80 °C and never refrozen, to avoid batch variability and aggregation that can suppress HDR efficiency. For guides, we’d use chemically synthesized sgRNAs (crRNA:tracrRNA duplexes), which provide improved stability, uniformity, and activity relative to in vitro-transcribed (IVT) sgRNAs 5–6. IVT sgRNAs frequently contain truncated species, double-stranded contaminants, and innate immune triggers 23. If IVT guides are used, they should be HPLC- or PAGE-purified 5.
A defining advantage of the landing pad architecture is that payload guide design is performed once. Because the synthetic homology arms and synthetic protospacers flanking the stuffer cassette are engineered sequences absent from the genome and every insert, the same sgRNA is reused for every swap, and HDR relies on invariant homology boundaries. These guides function as permanent “swap guides” for the platform. The synthetic protospacers are positioned so that Cas9 cut sites align with the internal synthetic homology arms, enabling reconstruction of precise junctions without residual stuffer sequence. Because both the boundaries and cut sites are fixed, correct HDR should produce the same junction sequence in every integration, allowing standardized quality control using left and right junction PCR rather than full-cassette sequencing.
To maximize editing efficiency, we propose assembling Cas9 RNP complexes fresh on the day of transformation. We’d combine Cas9 with sgRNA at an approximately 1:1.2 molar ratio and incubate briefly at room temperature to promote complex formation, after which we’d maintain the RNPs on ice until electroporation. Prior to electroporation, we’d pass complexes through a 0.22 µm low-protein-binding filter to remove aggregates, further improving HDR efficiency.
Cell preparation and transformation
The balance between HDR and NHEJ in C. reinhardtii is strongly cell cycle-dependent, with HDR frequency peaking when synchronized cultures re-enter the light in a near-uniform G2/M-like state following dark cycling 4. Accordingly, the landing pad system is built around a standardized transformation regime that maximizes HDR competence and minimizes uncontrolled variability across experiments. Cultures should be synchronized using two 12 h 28 °C light → 12 h 18 °C dark cycles (48 h total), and electroporation should be performed 12 h after the onset of a third light period, under continuous cool-white LED illumination at ~100 µmol photons m−2 s−1, as previously described 4. This synchronization strategy should enrich for HDR-competent cells and provide a reproducible temporal window for Cas9 activity.
Because CC-124 and most other swimming-competent laboratory strains retain an intact cell wall, transformation efficiency and precision genome editing require controlled cell wall removal prior to electroporation. For this purpose, we adopt an autolysin-based wall digestion strategy, which has been shown to be both necessary and sufficient for efficient CRISPR-Cas9-mediated knock-in editing in walled Chlamydomonas strains 6. In this approach, synchronized cultures are treated with gamete-derived autolysin to achieve near-complete wall removal, followed by osmotic stabilization and electroporation with Cas9 RNPs and highly purified, linear donor DNA. Complete wall removal is a key determinant of both transformation efficiency and HDR fidelity; incomplete digestion substantially reduces correct integration frequency and increases stochastic outcomes 6.
We propose using electroporation parameters adapted from internal protocols (~500 V, 50 µF, 800 Ω) in a BioRad Gene Pulser Xcell Electroporator with a 4 mm cuvette, with cells, gel-purified, linearized donor DNA, and Cas9 RNPs kept on ice until pulsing. Following electroporation, we’ll let cells recover in TAP medium supplemented with sucrose and incubate in the dark overnight to minimize metabolic stress and support DNA repair 4–5. These transformation and recovery conditions are deliberately standardized across the entire platform. We’ll apply them with high rigor during one-time landing pad installation (Stage 1) to maximize HDR fidelity and long-term genomic stability, and reuse them unchanged during high-throughput payload swapping (Stage 2) to ensure that observed differences between GOI and variants reflect biological signal rather than transformation-induced variability.
Strain-building and quality control workflow
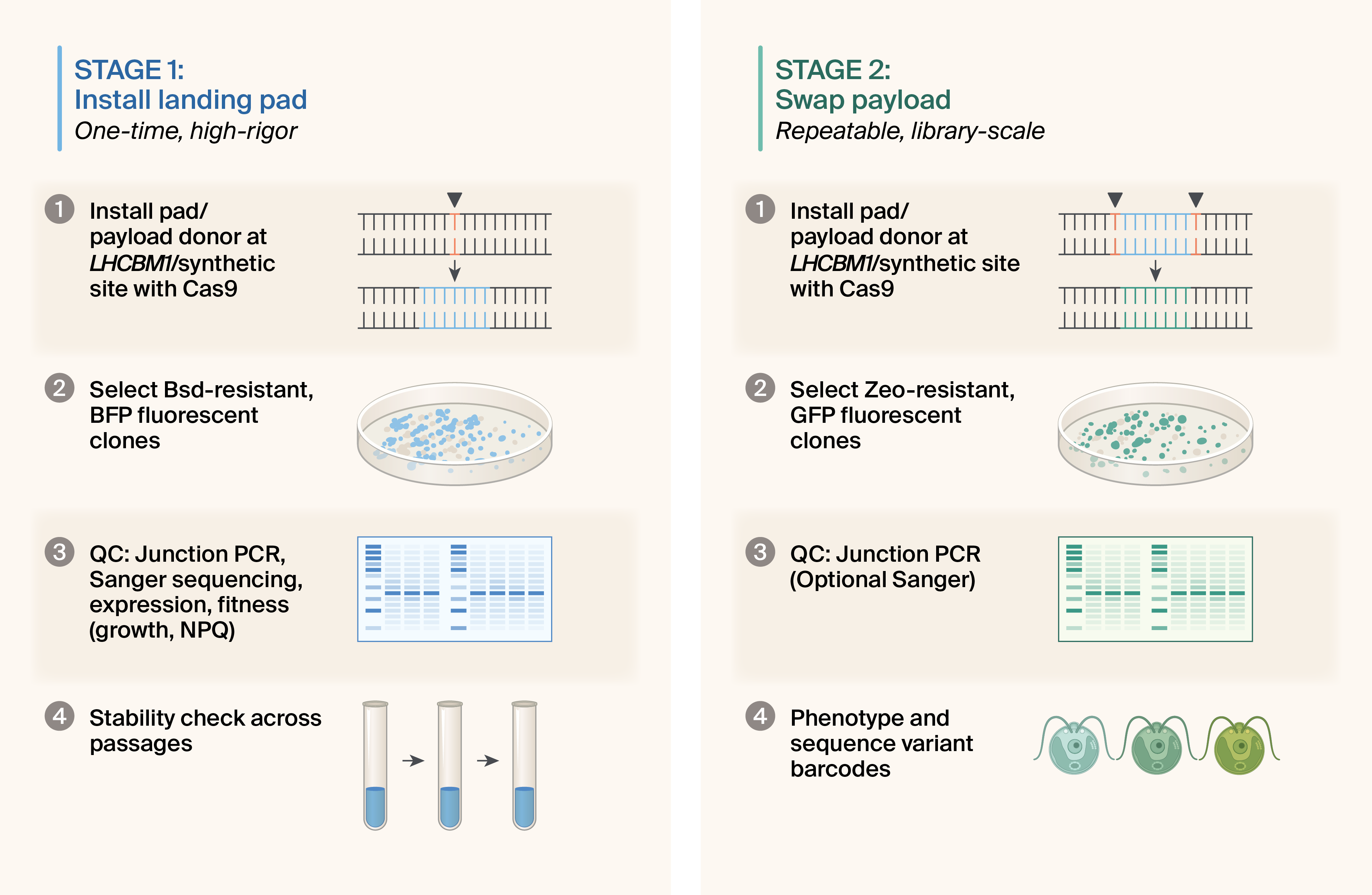
Figure 2. Landing pad installation and implementation workflow.
Stage 1 (left): One-time installation and validation of the landing pad at the LHCBM1 safe-harbor locus in a srta-attenuated CC-124 background. Following Cas9-mediated HDR insertion, candidate lines undergo high-rigor quality control, including verification of stuffer cassette function (BFP fluorescence and blasticidin resistance), left and right junction PCR and Sanger sequencing, growth and photophysiology assays, and extended passaging to confirm long-term expression stability. Lines that meet all criteria are banked as reusable chassis strains.
Stage 2 (right): Repeatable, high-throughput payload swapping. Cas9 RNPs excise the stuffer cassette at synthetic protospacer–PAM cut sites, and a linear donor template installs the desired payload by HDR. Correct integrants are enriched by loss of stuffer markers (BFP-negative, blasticidin-sensitive) and gain of payload selection (e.g., zeocin resistance and/or payload fluorescence), followed by standardized junction PCR and optional spot-check sequencing. This stage can be iterated to install single constructs or variant libraries in a fixed chromosomal context.
The logic of the landing pad is built around a simple architectural separation: Stage 1 is slow, high-rigor, and performed once; Stage 2 is fast, QC’d in bulk, and repeatable at scale (Figure 2).
Stage 1: Building the universal landing pad strain
Using the standardized transformation conditions described above, we propose installing the landing pad as a one-time genomic modification that defines the foundation for all subsequent payload swaps. We’ll perform installation with a linear donor flanked by ~50 bp of genomic homology downstream of the LHCBM1 locus. The donor encodes the complete landing pad architecture: an HSP70A-RBCS2 promoter, RBCS2 intron 1, aadA terminator, engineered synthetic homology interfaces, synthetic protospacers, and the fluorescent protein plus blasticidin-resistance (FP and BsdR) stuffer cassette.
For this initial installation, we’ll target Cas9 to the endogenous LHCBM1 locus using one sgRNA that cuts within a non-coding region immediately downstream of the gene. Our proposed guide placement avoids the promoter, coding region, and annotated 3′ UTR. Under these conditions, a single Cas9-induced double-strand break should enable complete HDR-mediated insertion of the full landing pad cassette in one step 4–6. These “installation guides” are designed to be used only once to build the chassis and are conceptually distinct from the synthetic swap guides reused during payload replacement.
Because this installation determines the behavior of every future payload, the resulting strain should be subjected to comprehensive, high-rigor validation drawn from established C. reinhardtii genome-editing and safe-harbor integration workflows 4–6, 8. Validation should include:
- Left and right junction PCR
- Sanger sequencing across both junctions to exclude partial HDR or plasmid-backbone artifacts
- Fluorescence imaging or FACS to confirm robust stuffer FP expression
- Blasticidin selection to confirm stuffer resistance, with optional qPCR to quantify transgene expression
- Growth rate and chlorophyll/photophysiology measurement to assess potential fitness perturbations
- LHCBM1 qPCR to ensure native gene expression remains within acceptable limits 8
- Stuffer qPCR to confirm concordance between transcript abundance and fluorescence
Expression stability should then be evaluated by extended passaging. Although SRTA-mediated repression is strongly reduced in the chassis background, additional epigenetic pathways can still contribute to gradual expression decay over time 7, 14. Candidate lines should therefore be passaged for 15–20 generations under varying light intensities, with FP fluorescence and blasticidin resistance monitored as functional readouts of sustained transcriptional activity. Only lines that maintain stable expression across these perturbations qualify as universal landing pad strains. Once a strain clears this validation pipeline, we will bank it as the standardized chassis for Stage 2 high-throughput payload swapping. Whole-genome sequencing at this stage can be employed to confirm correct insertion structure and audit for unintended integrations in selected chassis lines 4–6.
Stage 2: High-throughput transgene payload insertion
Each payload insertion can be implemented as a drop-in swap of the stuffer cassette using a standardized HDR workflow, in either single-plex or pooled modes. In single-plex swaps, a single donor installs a single payload, allowing us to screen and characterize clonal isolates directly. In pooled swaps, a mixed donor library will install many payload variants in a shared culture, and we can quantify variant representation by sequencing barcode abundance over time or across selection conditions. Cas9 RNPs should cleave the synthetic protospacer sequences flanking the stuffer, and we’ll supply a purified, linearized donor template to repair the resulting gap by HDR at the synthetic homology arms. Colonies (or FACS-isolated cells) that become simultaneously FP-negative and blasticidin-sensitive should be enriched for full stuffer replacement. We expect partial HDR events, NHEJ-mediated small indels, and misrepair to retain at least one intact stuffer marker and should therefore become depleted at this stage. Correct payload insertion will confer zeocin resistance and drive tag expression (fluorescent or otherwise); zeocin selection provides strong biological amplification of rare HDR events, while fluorescence screening (imaging or FACS) can confirm proper expression. This combined double-negative (stuffer loss), double-positive (payload gain) phenotype enables rapid, high-throughput quality control. In pooled swaps, we’d apply this QC at the population level: zeocin will enrich for payload-containing integrants, we’ll verify loss of stuffer markers (BFP negativity and blasticidin sensitivity) by flow cytometry/FACS or microscopy (and, where applicable, gain of payload fluorescence), and we’ll spot-check correct integration via junction PCR (left and right) on pooled genomic DNA (and, when relevant, orientation-specific assays), with occasional Sanger sequencing of sampled isolates.
For library-scale swapping, exhaustive sequencing of every variant is neither necessary nor practical. The landing pad architecture is designed such that the most common failure modes should self-report phenotypically through retention of stuffer markers or loss of payload selection. Accordingly, we’ll reserve Sanger sequencing for representative clones and spot checks of atypical or unexpected phenotypes. During initial development and validation of the payload backbone, however, we’ll perform more intensive QC to establish empirical confidence in downstream high-throughput use, including occasional whole-genome sequencing of representative integrants to audit for off-target insertions or unintended structural rearrangements. This mirrors the validation strategy proposed during landing pad installation (steps 1–7), including fluorescent tagging and sequencing of multiple independent integrants per batch to estimate the frequency and spectrum of unintended polymorphisms.
Once this baseline is established, subsequent payload swaps rely on a reduced, standardized diagnostic set:
- Left and right junction PCR
- Diagnostic restriction mapping: Synthetic restriction sites embedded in and around the payload provide a rapid digest-based readout to distinguish correct from aberrant insertions.
- Barcode confirmation: The non-transcribed barcode region downstream of the terminator serves as a stable identifier confirming payload presence, orientation, and identity in pooled assays.
- qPCR measurement of LHCBM1 and payload expression: Used to verify that payload insertion leaves the local transcriptional environment intact and produces expression within the expected range.
QC is tiered: cheap, phenotype-based screens should eliminate most incorrect clones up front, and only a small fraction proceed to more detailed assays. For pooled libraries, PCR and sequencing can be applied to pooled genomic DNA, enabling scalable monitoring of library integrity without clone-by-clone handling.
Failure modes and mitigations
The architecture is explicitly designed such that common failure modes are either suppressed or rendered phenotypically observable.
Silencing (SRTA-mediated and residual pathways): The SRTA pathway selectively targets transgenic DNA for epigenetic silencing in C. reinhardtii, particularly when foreign promoters or heterologous coding sequences are present 7. Our chassis incorporates SRTA disruption, which should eliminate the dominant axis of transgene-specific silencing at its source. Nevertheless, additional epigenetic pathways can contribute to gradual expression instability over time 14. To mitigate residual silencing pressures, we retain an endogenous-like expression architecture (HSP70A-RBCS2 promoter with RBCS2 intron 1), which has been shown to be intrinsically more stable and resistant to repression than strongly heterologous promoter-ORF combinations 7, 14. This layered strategy should reduce both acute and long-term silencing risk without relying on any single mechanism.
Locus dependency and chromatin variability: Random genomic integration in C. reinhardtii routinely produces 10–100× variation in transgene expression, obscuring functional differences across variant libraries 6, 8. By anchoring all payloads to the LHCBM1 locus, a validated nuclear safe harbor, our proposed system removes locus-dependent chromatin variability as a confounding factor 8. As a result, expression differences between variants should primarily reflect payload-intrinsic properties rather than positional effects.
NHEJ dominance and failed HDR: Even under synchronized conditions, homology-directed repair must compete with NHEJ 4–5. This system mitigates NHEJ outcomes in two complementary ways. First, we try to bias repair toward HDR through the use of linear donor templates and high-quality Cas9 RNPs, which is standard practice for efficient nuclear editing in C. reinhardtii 4–6. Second, NHEJ outcomes should be phenotypically obvious by design. If Cas9-mediated cleavage is repaired by simple re-ligation of the stuffer cassette, colonies remain FP-positive and blasticidin-resistant and are discarded during selection.
Partial HDR and remnant stuffer sequence: Partial HDR events are structurally disfavored by design. Because complete loss of both fluorescence and blasticidin resistance requires removal of the promoter, intron, and initial coding elements of the stuffer cassette, any incomplete repair leaves at least one functional marker intact. Such clones are automatically excluded by the dual-negative screen. Sharp synthetic boundaries and short homology arms should further reduce microhomology-mediated repair and aberrant junction formation 5–6.
Off-target integration and tandem insertions: Ectopic integration of donor DNA via NHEJ, including tandem insertions or off-target payload copies, is a known risk in nuclear transformation. However, such events are unlikely to confer both loss of the stuffer markers and acquisition of zeocin resistance in the absence of correct HDR at the landing pad. Clones with off-target insertions but intact stuffer cassettes are therefore eliminated during selection. For the universal landing pad chassis, we can use whole-genome sequencing to audit for unintended integrations. During high-throughput payload swapping, linear donors, RNP delivery, and phenotype-based filtering should further suppress rare off-target events, and we can monitor such events by spot-check sequencing or pooled PCR-based assays as needed 4–6. If it's important to rule out rare “correct swap and extra copy” outcomes, either ectopic insertions elsewhere in the genome or tandem concatemers of the payload donor at the landing pad, we can screen representative integrants for payload copy number by qPCR/ddPCR (payload ORF or shBle normalized to a single-copy nuclear reference), with occasional whole-genome sequencing audits as needed. We can also flag major unintended perturbations by routine growth and photophysiology benchmarking.
On-target misrepair and donor capture: In addition to failed HDR and ectopic insertion, a minority of outcomes can arise from on-target NHEJ capture of the donor at the landing pad locus. These include incomplete stuffer replacement, insertion of the payload in reverse orientation, or junction architectures that duplicate portions of the synthetic homology arms. Such events should largely be filtered by the dual-marker logic: retention of stuffer fluorescence or blasticidin resistance flags incomplete replacement, while correct swaps regenerate the expected junctions at both homology boundaries. When structural uniformity matters, we can detect these rare outcomes using left and right junction PCR (and, where relevant, orientation-specific PCR spanning promoter-to-ORF or ORF-to-terminator), with occasional Sanger sequencing of junction amplicons. Although some junction variants may remain functional, duplicated homology or inverted architectures are excluded when long-term stability or iterative reuse is required.
UTR-driven misexpression and steric effects: Uniform promoter-intron-terminator architecture across all payloads should eliminate UTR-driven expression variability. For proteins sensitive to C-terminal fusion, the small-tag backbone should minimize steric interference by using compact epitope tags, while fluorescent-tag backbones remain available when localization or expression visualization is required.
Library noise and misassigned variants: Payloads incorporate a non-transcribed barcode region that should be genetically stable and unaffected by promoter dynamics or protein localization. In pooled experiments, barcodes should remain faithfully linked to their corresponding variants, enabling robust quantification of variant fitness or enrichment even when some coding sequences are under strong selection.
Future vision
Multi-pad C. reinhardtii for multi-gene engineering: The architecture is inherently expandable. Just as one pad can host hundreds of variants, two or three pads placed in different safe-harbor loci would enable combinatorial expression of multiple genes at defined stoichiometries. DNA-free co-targeting can edit two nuclear genes simultaneously at ~22% efficiency in C. reinhardtii, enabling multi-locus designs 13. Inclusion of an additional unique CRISPR cut site and/or recombinase landing pad within the payload would enable iterative payload addition as an alternative to payload swapping, circumventing the reduced efficiency of CRISPR-mediated HDR for large DNA fragments by distributing integration across multiple events.
Promoter tuning and modular expression-strength panels: Because the pad decouples transgene expression from locus effects, promoter benchmarking is possible. We can construct standardized libraries of promoter variants (constitutive, inducible, and environmentally responsive) and test them in identical chromosomal contexts. Future landing pad designs could allow the promoter to be replaced independently of the payload or integrated as a parameterized module in the donor backbone. This would enable titration experiments, conditional rescue, and systematic exploration of dosage — distinguishing copy-number effects from regulatory and protein-intrinsic constraints.
CRISPRa/CRISPRi docking: As CRISPR activation and interference systems mature in algae, the landing pad can facilitate the installation of these transcriptional regulators. By embedding operator sequences or recruiting domains within pad boundaries, we can build inducible, tunable, or condition-responsive expression platforms for precise temporal control. This enables pulsed expression of toxic proteins, transient silencing of essential genes, and rescue experiments in knock-out contexts.
Barcode atlas for pooled screens: The barcode region in the payload is a placeholder; short, simple barcodes could be employed, or more designed barcode blocks. A standardized barcode block flanked by constant primer sites would enable pooled screening with direct NGS barcode quantification. Barcodes would either be pre-assigned for arrayed libraries or decoded once for pooled donor pools, establishing a stable barcode-to-variant map. QC for each payload would include verifying barcode presence and size during donor assembly, ensuring accurate downstream tracking, and enabling high-throughput variant screening and deep mutational scanning.
A generalizable framework for new Zoogle species: The landing pad concept described here is portable to other unicellular algae and diverse protists. In the context of the Zoogle discovery pipeline, which seeks non-model organisms with compelling biology, once CRISPR transformation methods are established, the following workflow could be followed: identify a permissive chromatin region; install a minimally functional landing pad; stabilize expression across passages; and thereafter bootstrap variant libraries and synthetic gene systems. Safe-harbor logic, synthetic homology, and standardized QC extend naturally to new chassis as they're discovered.
Final thoughts
By establishing LHCBM1 as the canonical nuclear safe-harbor locus, we anchor engineering to a part of the genome with predictable chromatin, high expression capacity, and proven tolerance for insertions 8. By placing a robust promoter-intron-terminator structure there, we insulate expression from environmental fluctuations and silencing pathways that disrupt most foreign sequences 7. By embedding synthetic CRISPR boundaries and homology arms into the architecture, we convert each swap into a standardized, junction-defined event rather than an uncontrolled competition between HDR and NHEJ 4–6. And by including a dual-marker stuffer cassette, we enable universal, phenotype-driven QC readout.
All of these elements reinforce one another, and none are individually novel. The challenge of implementation at scale lies in the initial development, execution, and validation of the landing pad and insert designs. However, once complete, this should enable high-throughput knock-in, in turn facilitating experiments such as profiling mutational landscapes of photosynthetic enzymes, screening proteins via swimming and phototaxis, refactoring pathways across chloroplast–nuclear boundaries, and designing genetic circuits.
Pub preparation
We used ChatGPT (GPT-5.1 and GPT-4o) to write text that we edited, expand on summary text that we provided and then edited the text it produced, help clarify and streamline text that we wrote, help copy-edit draft text to match Arcadia’s style, suggest wording ideas and then chose which small phrases or sentence structure ideas to use, and rearrange text to fit the structure of one of our pub templates. ChatGPT also suggested papers on relevant science, we did further reading, and we cited some of this literature. We used Cursor (GPT-5.1 codex and composer) to draft preliminary Python scripts for figure SVG mock-ups to spatially brainstorm, though we created final figures de novo in Adobe Illustrator.
Weigh in!
To guide development of the landing pad chassis and ensure community alignment, we invite feedback on a small number of design choices that remain intentionally configurable in early implementation. We’re not looking to freeze the architecture prematurely, but aim to stress-test components for reuse, scalability, and adoption across different C. reinhardtii workflows, while keeping the initial release as simple as possible.
Because the landing pad serves as a long-term genomic interface, early feedback has a multiplicative effect: choices made now (at v1) will shape what's easy or hard to do for years afterward. We're particularly interested in both reactions to the incremental development approach itself and in how researchers envision ultimately using this system.
Importantly, we can incorporate feedback at multiple layers — payload design, barcode format, promoter modules, iterative swapping behavior, and prioritization of additional safe-harbor loci — without altering the architecture’s fundamental logic. Input from researchers working across algal genetics, photosynthesis, optogenetics, protein engineering, genomics, and synthetic biology is therefore especially valuable at this stage.
1. Payload expression architecture
Should the default backbone emphasize:
- Fluorescent-tagged payloads (facilitates QC, localization, expression monitoring)
- Minimal-tag payloads (maximizes functional neutrality)
- A dual-release architecture with both formats available (e.g., prioritizing neutral functional assays vs. imaging-first workflows)
2. Barcode strategy for pooled screens
For pooled applications, should v1 adopt:
- A minimal 12–20 bp barcode block (current design)
- A complete standardized barcode module (longer, error-correcting, flanked by constant primer sites) that enables ready-to-use pooled fitness assays
3. Iterative replacement vs. locus sealing
Payload donors can either:
- Reintroduce protospacer–PAM modules, enabling iterative round-by-round replacement
- Omit them, permanently sealing the locus after insertion
Which mode should the default payload backbone support? Should both donor types be provided (should v1 privilege long-term reusability or genomic finality by default)?
4. Promoter and terminator variants
How important is tunable expression? Should additional regulatory modules be included in a v1 expression panel?
5. Expansion to multi-pad architectures
Is there desire for early exploration of additional safe-harbor loci to support:
- multi-gene circuits
- pathway refactoring
- stoichiometrically tuned expression
Or should v1 remain focused on a single optimized site?
6. Anything else?
Are there additional use-cases, constraints, or failure modes that would meaningfully affect the short- or long-term utility of a C. reinhardtii landing-pad system?
Be the first to comment on this publication.